

Recevez notre brochure sur les monte-escaliers
- Déjà demandé plus de 15 000 fois
- Trouvez le monte-escalier qui vous convient
- Prenez le temps de décider
« * » indique les champs nécessaires

SMA est l’acronyme utilisé pour désigner l’amyotrophie spinale. C’est une maladie qui affaiblit progressivement les muscles, jusqu’à ce qu’ils ne fonctionnent plus. Plus la maladie se développe tôt, plus les muscles se détériorent rapidement. Le plus souvent, les symptômes surviennent chez les jeunes enfants. Chaque année, environ 10 enfants sur 100 000 en sont atteints. Pour eux, Il y a de bonnes nouvelles ! L’un des médicaments disponibles vient d’être approuvé par l’Agence européenne des médicaments. Et un deuxième médicament est à l’étude.
L’amyotrophie spinale est une maladie neuromusculaire : il y a un défaut dans les cellules nerveuses qui transmettent un signal du cerveau aux muscles. Nous appelons ces cellules nerveuses des « motoneurones ». Si les muscles ne reçoivent pas de signal de la part des motoneurones, ils ne répondent pas et ne bougent donc pas. Les muscles qui ne font (presque) rien s’affaiblissent et s’atrophient jusqu’à ce qu’ils ne fonctionnent plus du tout. Les muscles finiront par être paralysés. La maladie musculaire SMA affecte principalement les muscles des bras et des jambes. Mais à mesure que la maladie progresse, les muscles respiratoires sont également touchés. Le corps ne reçoit alors plus assez d’oxygène. Cela peut nécessiter une assistance respiratoire chronique à l’aide d’un appareil.
L’amyotrophie spinale est causée par une erreur dans l’un des quelques 20 000 gènes que possèdent les êtres humains. Tous ces gènes sont des morceaux d’ADN qui déterminent à quoi ressemble une personne, qui elle est et si elle est en bonne santé. Les gènes sont situés sur les chromosomes ; chaque être humain en possède 46 (23 paires). Les scientifiques ont découvert que la cause de l’amyotrophie spinale se trouve sur le 5ème chromosome. Chez les personnes atteintes de SMA, le gène SMN1 a un défaut ou est totalement manquant. Ce gène produit normalement la protéine SMN, qui est le carburant des motoneurones. Les motoneurones ne peuvent pas fonctionner (correctement) sans cette protéine : les signaux du cerveau ne sont pas transmis correctement ou pas du tout aux muscles. Chez les personnes atteintes de SMA, le gène SMN2 (une sorte de « gène de réserve ») prend en charge la tâche de fabriquer la protéine SMN. Mais le gène SMN2 ne produit que 10 % de la protéine utile produite par le gène SMN1.
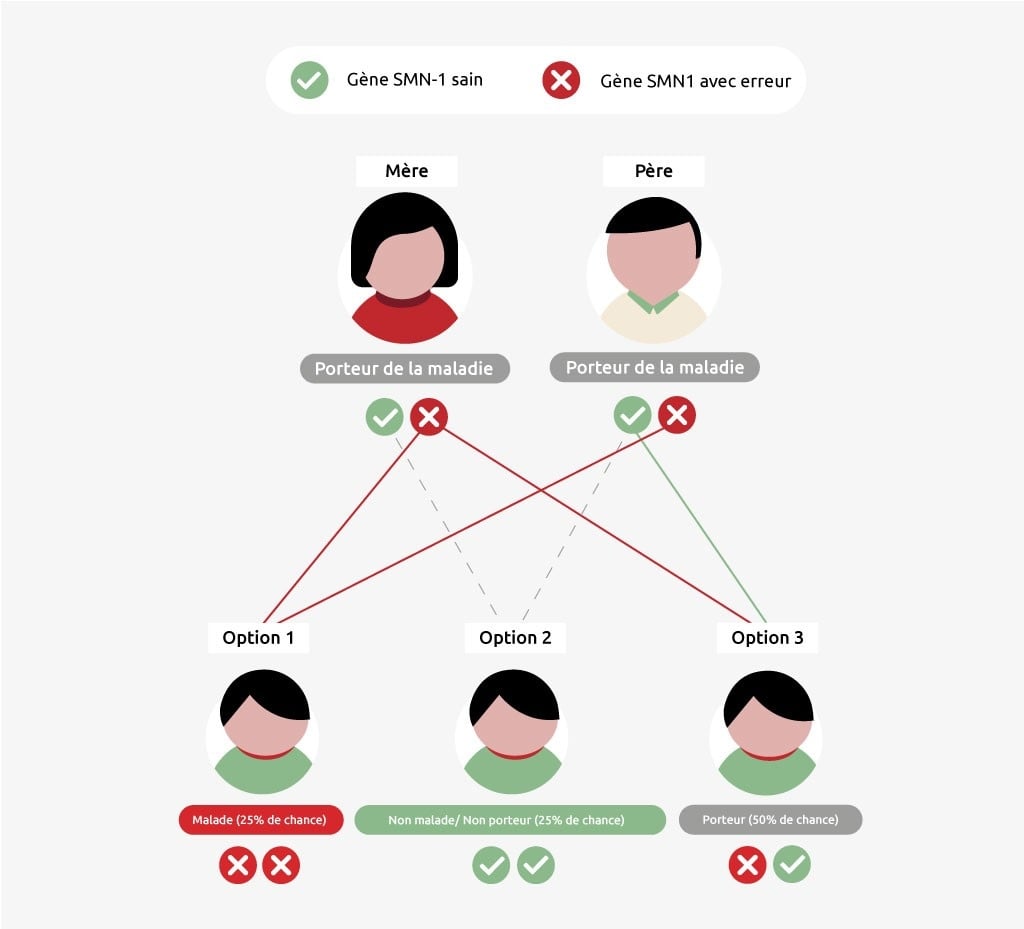
La SMA est une maladie héréditaire : ce sont les parents qui transmettent les gènes défectueux à leurs enfants. Si un parent est porteur de la maladie, il a un gène SMN1 sain et un gène défectueux. Un porteur n’est pas lui-même malade, mais peut transmettre la maladie. Les enfants ne contractent la maladie SMA que si leur père et leur mère leur transmettent un gène SMN1 défectueux ou un 5e chromosome sans gène SMN1. La probabilité que les enfants de deux porteurs tombent malades est de 25 %. Celle que les enfants deviennent seuls porteurs est plus grande : 50 %. Ils ont également 25% de chances de ne pas hériter du tout de la maladie ou du statut de porteur.
Il existe 4 types d’amyotrophie spinale. La distinction entre les types est faite en examinant l’âge auquel la maladie se développe et la rapidité avec laquelle les symptômes s’aggravent. Parce que la maladie est souvent diagnostiquée à un jeune âge, la SMA peut être considérée comme une maladie des bébés et des jeunes enfants. Plus la maladie survient tôt, plus l’évolution est défavorable.
La différence dans l’évolution de la maladie est due au fait que certaines personnes ont plus de « gènes de réserve » du gène SMN2. Plus quelqu’un a de ces gènes, plus il peut produire de protéines SMN et moins il manque de protéines SMN. Cela permet aux motoneurones de travailler plus longtemps. Quelqu’un avec 0 à 2 exemplaires développe le type 0 ou I. Avec 3 exemplaires, le type I, le type II ou même le type III. Avec 4 exemplaires ou plus, le type II (dans certains cas), le type III ou le type IV se développe.
La SMA de type I survient chez les bébés, juste après la naissance ou au cours de leurs 6 premiers mois de vie. Les muscles des bras, des jambes et du tronc sont souvent affaiblis. Les muscles respiratoires deviennent également plus faibles. Les bébés atteints de SMA n’apprennent généralement pas à s’asseoir, à tenir la tête haute ou à se retourner. Ce type est la forme la plus grave de la maladie. Ici aussi, plus la maladie se développe tôt, plus les symptômes sont sévères. Dans certains cas, la maladie peut se manifester avant la naissance, auquel cas elle est appelée SMA de type 0. Le type I survient chez environ 55 % des personnes atteintes d’amyotrophie spinale.
L’amyotrophie spinale de type II survient chez les enfants entre 6 et 18 mois. Ils souffrent principalement de muscles plus faibles au niveau des jambes et du dos. Les bras sont souvent encore relativement forts. Les bébés atteints de cette forme de maladie musculaire SMA peuvent développer la capacité à s’asseoir et à se retourner, mais ils ne peuvent pas marcher seuls. Les enfants et adolescents atteints de la SMA de type II ont donc presque toujours besoin d’un fauteuil roulant. Leur espérance de vie est comprise entre 10 et 40 ans. Cela peut donc varier considérablement d’une personne à l’autre.
Si la SMA se développe chez les enfants entre 18 mois et 4 ans, on parle de type III. Les enfants développent souvent la capacité à s’asseoir et à marcher de manière autonome. Cependant, il y a de fortes chances qu’ils perdent ces compétences plus tard parce que leurs muscles s’affaiblissent. Les enfants atteints de SMA de type III doivent donc souvent se déplacer en fauteuil roulant. Plus tard dans la vie, il leur sera peut-être nécessaire d’utiliser un appareil respiratoire. L’espérance de vie des malades atteints du type III diffère également. Les personnes diagnostiquées avec un SMA de type III ont généralement une espérance de vie normale.

L’amyotrophie spinale de type IV est très rare. Les symptômes ne surviennent qu’après l’âge de 30 ans. La plupart des personnes atteintes de ce type de SMA ne présentent que des symptômes bénins. Des périodes de dégradation de l’état de santé alternent avec des périodes sans symptôme. Les personnes atteintes de SMA de type IV ont souvent une espérance de vie normale. Cependant, à un âge plus avancé, ils peuvent avoir des difficultés à monter les escaliers et à lever les bras.
Dans la plupart des cas, l’amyotrophie spinale est diagnostiquée par des tests ADN qui permettent d’identifier des anomalies. Toutefois, le type de SMA ne peut pas être déterminé de cette manière, mais par l’observation du moment où la maladie se déclenche et de la vitesse à laquelle elle se développe.
Dans 8 % des cas, l’anomalie du gène ne peut être démontrée. Un autre type d’examen est alors requis : un examen électromyographique (EMG) ou une biopsie musculaire. Lors de l’examen électromyographique, de fines aiguilles sont insérées dans les muscles. Les aiguilles sont connectées à l’appareil EMG qui mesure les réponses des muscles. Le médecin peut alors voir si les muscles réagissent de façon anormale. Lors d’une biopsie musculaire, le médecin prélève un très petit morceau de muscle pour l’étudier au microscope. Le médecin examine la structure du muscle et effectue des tests. Par exemple, le médecin examine la taille des fibres musculaires. Si leur taille diffère beaucoup, le diagnostic de SMA est établi.
Plusieurs médicaments ont été développés pour traiter la SMA et en réduire les symptômes. Le médicament Spinraza est actuellement approuvé en Europe. Il est administré par péridurale, et permet au gène SMN2 d’être stimulé plusieurs fois par an de manière à produire une protéine SMN plus utile. Les motoneurones ont ainsi plus de carburant pour contrôler les muscles.
Il existe également un médicament, appelé Zolgensma, qui utilise la thérapie génique pour créer un nouveau gène SMN1 dans les motoneurones. L’avantage est que ce médicament ne doit être utilisé qu’une seule fois. Les inconvénients sont le coût du traitement, qui s’élève à 2 millions d’euros par patient, ainsi que le manque de clarté sur ses effets à long terme. En Europe, ce médicament est approuvé pour une utilisation chez les jeunes enfants. Mais des accords doivent encore être conclus sur le remboursement du médicament avant que son utilisation ne puisse être généralisée.
Plus sur la santé





